 Lindsay Cade
Lindsay Cade
Academia.edu Software Engineer & Hackbright Alum
Before her current software developer incarnation, Lindsay was a traveler, a research biologist and an illustrator. She is still all of those things to varying degrees but mostly writes code now as a software engineer at Academia.edu. Before Hackbright Academy, Lindsay earned her bachelor’s degrees in Biochemistry and Illustration from RIT.
By Lindsay Cade (Hackbright Academy – spring 2013 class)
Hackbright Side Project: Finds likely presence of insertion or deletion in each of a set of DNA sequences. Given the unaltered DNA sequence and two short sequences flanking the site where the mutation is expected to occur, app displays how far apart the flanking sites are while allowing for fuzzy matching of flanking sites and deciding the direction of the sequences (DNA sequences can be sequenced in either a forward or reverse direction).
Project developed in: Python, BioPython, Flask. Older version uses: R, BioStrings.
SCOPE OF PROBLEM
In my old lab, we (I) had to process a lot of DNA sequences. You know, something like this “AAGGCGATTAAGTTGGTTCCCAG”. They would come 96 at a time and I was looking for the presence of an indel, or insertion or deletion of DNA basepairs, in each one. Most didn’t have one, but those that did were considered a victory for our project.

When I was working in the lab, I frequently thought to myself that there MUST be a way for computers to solve this since it was comparing letters to other letters and that is something that computers excel at. At the time, though, I didn’t have any programming skills, so I continued manually slogging through pages and pages of sequences and then went home and made eyes at my programmer husband attempting to convince him that he should help me.
But why was matching and finding so hard? Shouldn’t it should be just a simple ctrl+F function to find sequences around the mutation sites? Easy peasy! But as it turns out, biology is complicated (who knew) and there were several complicating factors.
First, the things that were definitely common across the sequence set:
* They were (supposed to be the) same gene
* There was the presence of a known, common flanking site on each side of area where indel was expected. The area in which we expected the indel to be I will refer to as the spacer.

The process of sequencing DNA samples, though, is not perfect, and anomalies could have been introduced through several channels.
Each sequence may be or have:
* a variable length,
* a variable indel length,
* unclean sequencing (bases showing as N’s instead of ATCG),
* polymorphisms (sometimes a letter in the sample DNA would be switched in a large percentage of the sequences and while this is sort of normal and OK, it makes simple find functions hard),
* a reverse complement (if you’re not sure about this concept, see Complementarity on wikipedia).
The goal of going through all these sequences was to get a few lines to put into a journal paper — listing the wild type sequence (or, the expected un-mutated sequence) and the insertions and deletions of the set of sequences aligned against each other showing the differences. Like this:
PROGRAM OUTPUT
Now that I’ve cut my programming teeth at Hackbright, I was able to write an automated solution to this problem fairly easily with Python!
In my implementation, a user inputs:
* two flanking sites
* the sequences to be analyzed (straight from the sequencing results website)
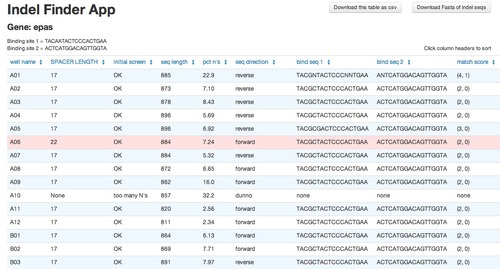
* the wild-type sequence
and out comes a table listing the distance between the flanking sites (below)! There are also several other columns with supplemental data so a user can trace through the steps and manually verify the program’s results in case of an error. My previous boss didn’t always trust computers 🙂
You can see that in the above example most sequences have a spacer length of 17, which is the wild-type spacer length. But A6 has a spacer length of 22, implying there is likely a 5 base pair insertion. Hooray!
The other columns provide the following:
* Initial screen: An initial look at each sequence to make sure they were long enough to be useable and to make sure it was “clean” enough to be worthwhile to interpret (i.e. not too many N’s)
* Sequence length: self explanatory
* Pct n’s: The percent of the bases in each sequence that was N’s
* Seq direction: see above
* Bind seq 1 and 2: shows the actual sequence of each flanking site that was chosen as the best position. This is for the case that something needs to be verified by hand. This will make it easier to find what the program is referring to in each sequence.
* Match score: see above
The table is sortable on each column so the whole sequence set can be explored at once and one can get an overview of the qualities of this sequencing run.
The program also outputs a FASTA formatted file in the browser containing the sequences with a spacer length different than the wild-type spacer length, all converted to the forward direction, that can be copied and pasted directly into an alignment app (we used EBI ClustalW2).
This app, when fed the FASTA file generated by my program, outputs text in the format needed for the journal papers.

HOW IT WORKS
So, how does this program solve the problem of messy biology and fuzzy matches?
Since a basic find function and regular expressions won’t work, I had to find another way to figure out where the flanking sequences were. Once I found the start and end index of each flanking site, it was easy to calculate the distance between them and infer the presence or absence of an indel.
The solution my Hackbright instructor and I came up with was to treat a sequence like a line and create a window, the length of the flanking sequence, that would “slide” along the sequence. At each position, the bases in the window are compared to one of the wild-type flanking sequences. For each mismatch between the wild-type flanking sequence and the sequence in the window, the score for that window would be increased by one. Therefore a perfect match would have a score of 0 and a window with no matching bases would have a score equal to the length of the flanking site.
This generates a score for each position in the sequence. The index of the lowest score would be the starting position for the flanking site.
This was easy to implement and worked well! When the results from the program are returned, they match almost perfectly with the manually found indels.
The list of scores, recorded in the far right column, derived from the “sliding window” allowed me to easily find the starting index of each flanking site. From there it was simple to calculate the difference between the two and compare that to the wild-type distance.
Another challenge was figuring out whether a sequence was forward (like the wild-type) or the reverse complement. The Biopython library makes it easy to calculate the reverse complement of sequences, so each flanking sequence was tested against the forward and reverse complement of each sequence. The direction in which the scores of both flanking sites was lowest was recorded in the direction column. If the scores from each direction were the same or conflicting, the sequence was discarded from the analysis. This doubled the time and space necessary for the computation, but seemed to work well.
POSSIBLE IMPROVEMENTS
Speed it up: The program runs a bit slowly in it’s current form. I tried pretty hard to make most things happen during one loop (the “sliding window” being the obvious exception) but it still takes a while. I’m sure there could be improvements.
Make the final alignments in the app: The app now requires copying and pasting text into another browser window where alignment can be done, but wouldn’t it be better if the program would just do it for you? The answer is yes. A couple of possible ways this could happen are with the Clustal Omega Web API, Clustal Omega offline tool, or the Python AlignIO package.
Tighten up indel detection: Occasionally a result will appear where an indel is over 100 basepairs long. This could happen but is very unlikely. The problem is caused by the lax rules when choosing the best score position, however these type of errors are easy to pick out by a human so I considered the program useable even with this bug.
Trim reversed sequences for alignment: Sometimes when a sequence is reversed, the placement of the flanking sequences is in such a different location relative to the whole sequence that when run through an alignment program, it looks like there is no match. To solve this, a user can usually just delete 50-100 characters off the front of the problem sequence, but that is not an elegant programming solution. With a bit of clever math this could be solved without any user intervention.
If you have any other suggestions, I’d be happy to hear them!
The code can be found on GitHub here.
This blog post was originally posted at Lindsay Cade’s blog.